c .上样染料和缓冲液选择
制备凝胶电泳时,样品中添加了凝胶上样缓冲液 (通常是6X或10X的原液)。 上样缓冲液包括如下组分:
密度成分,如甘油或蔗糖,增加样品粘度,确保样品沉入孔中。
盐,如Tris-HCl,为样品创造良好的离子强度和pH值环境。 高盐浓度的上样缓冲液会产生更大片或扭曲的条带以及污点。
金属螯合剂,如EDTA,可阻止样品中的核酸酶降解核酸。
染料 为监测样品的上样、电泳进展和pH值变化提供了颜色指示。 部分上样缓冲液可能包含多个染料,以便有效跟踪样品中不同大小分子的迁移。
通常,上样染料都是带负电荷的小分子,因此迁移方向与核酸相同。 其中有的显示pH值颜色,上样和运行时可作为样品的pH指示剂(图9A)。 常用染料包括溴酚蓝、二甲苯青,苯酚红,和橙色G。选择上样缓冲液时,需注意染料的表面迁移(s)(图9B, 表5和6)以避免遮盖目的核酸条带,特别是分子大小接近时(图9C)。 染料遮盖会影响目的条带的分析和量化,导致结果不可靠。
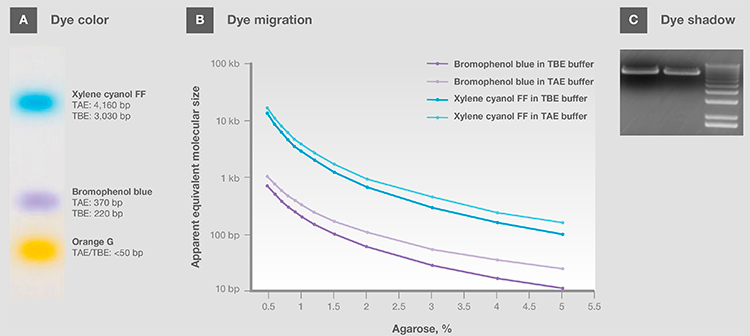
图9.(A)中性pH中染料颜色。(B)在TAE和TBE缓冲液中不同百分比琼脂糖里的染料迁移。(C)在可视化过程中染料阴影遮盖条带。
有时,上样缓冲液包含清洗剂或还原剂,如SDS, 尿素和甲酰胺,以用于变性。此类添加剂可破坏核酸分子内部和分子间的相互作用,促成线性或单链的分子构象。为获得最佳分离结果,应将样品和变性剂加入至上样染料中一同加热(图 10A)。对来源于酶反应的双链DNA电泳,可将SDS添加到上样缓冲液,以破坏蛋白质和核酸之间的相互作用,防止样品迁移性改变(图10B)。
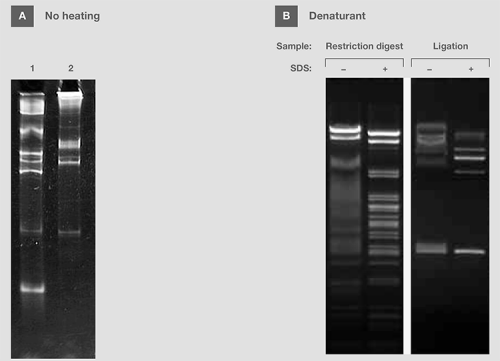
图10,热量、SDS对样品电泳的影响。(A) 在不加热处理的情况下,将含有RNA分子量标准的变性缓冲液加入凝胶中。 (B) 在有或无SDS的上样缓冲液中准备来源于限制性内切和连接反应的DNA样品。 在凝胶上样前,加热SDS中的样品。


3.运行电泳
在凝胶、标准品和样品制备后,进行电泳。拆卸梳子和添加电泳缓冲液前,凝胶须完全凝固。凝胶梳应平稳地向上提起,以免撕裂凝胶、扭曲胶孔。移除梳子和添加缓冲液后,注意清除孔里的气泡。对于聚丙烯酰胺凝胶,应用缓冲液彻底冲洗胶孔,以清除残留未聚合的丙烯酰胺。
水平凝胶应朝向凝胶盒,样品孔位于负极一侧,以便在电泳启动后,将样品移动至正电极侧(图11A)。该方向可记为“跑向红色”,因为正电极通常为红色。 垂直凝胶盒中,胶孔设计在顶部(图11B)。
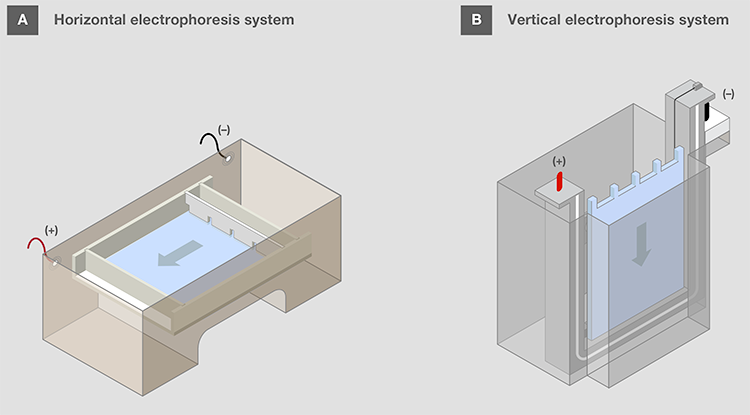
图11.水平和垂直电泳系统的凝胶装置。 箭头表示电泳中核酸迁移的方向。
a.电泳缓冲液选择
电泳缓冲液为具有缓冲能力的离子溶液,通常在凝胶中使用,以保证电流流动同时防止pH值变化。电泳过程中,由于电子流动,负极逐渐偏向碱性,正极逐渐偏向酸性,从而导致水电解和pH值改变(表6)。氢气和氧气的释放会引起电极起泡,这是凝胶运行的迹象。理想情况下,电泳缓冲液和凝胶制备缓冲液应相同,以确保有效的导电性。
表6.两个电极的化学反应和pH值变化。

电泳缓冲液的选择取决于样品大小、运行时间和后电泳过程,其中Tris-醋酸盐EDTA(TAE)和Tris-硼酸 EDTA(TBE)是最常用的两种缓冲液(表 7)[2,7]。
因为不容易形成污点,TAE适用于>1500bp的片段。由于缓冲能力较低,TAE更适合短时间的电泳运行(例如<2小时)。TAE缓冲能力较低,长时间运行凝胶会导致过热,样品变性和/或扩散以及凝胶融化(可能)。
TBE缓冲能力较高,不易导致过热,因此更适合长时间运行。对于较短片段的分离,TBE效果更佳,而在TBE中,dsDNA迁移得更慢。TBE可抑制酶,因此不适合涉及酶学步骤的下游应用,如限制内切、克隆和PCR。
使用离子强度高于1X TAE或0.5-1X TBE缓冲液,可更快地移动样品,但由于高导电性会产生大量的热量,容易导致样品变性和凝胶损伤。

凝胶应完全浸在缓冲液中,以确保离子流动,防止凝胶干(图12A)。然而,当使用水平凝胶时(例如,琼脂糖),凝胶上的缓冲液深度应低于3–5mm。缓冲液过高(>5 mm)会导致核酸迁移性降低,加大条带扭曲和过热。
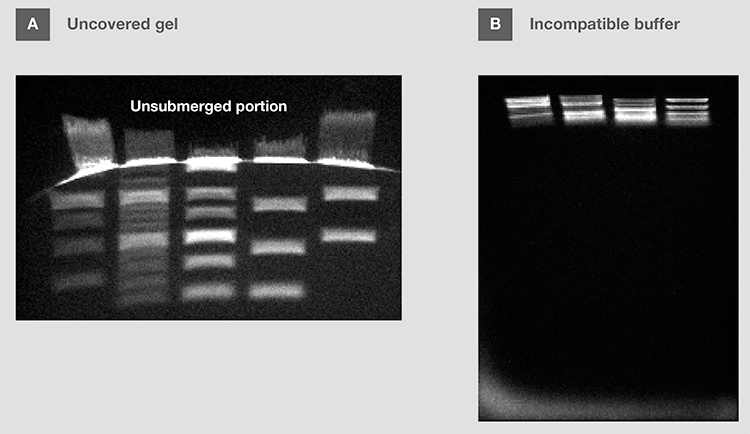
图12.电泳缓冲液对电泳的影响。(A) 在电泳过程中,凝胶的顶部部分未被浸没。(B) 在低于推荐浓度的盐浓度缓冲液中运行凝胶(与凝胶制备中使用的缓冲液不同)。
注意,变性琼脂糖的电泳缓冲液可能既不是TAE,也不是TBE,因为这些凝胶可能在不同的缓冲液中制备,如磷酸钠和MOPS。选择与所用凝胶兼容的电泳缓冲液非常重要(图12B)。
b.电压
采用恒定电压、电流或功率通过凝胶,施加电势,开始运行凝胶。核酸电泳中常用恒压,一般为5–10 V/cm。
施加的电压(V)=电极间的距离(cm)x建议V/cm
可根据需要分离的DN**段的大小调整电压,也可以根据所使用的电泳缓冲液的类型进行调整(表 8)。市售核酸分子量标准通常提供建议电压,以便每个产品片段实现最佳分离。注意,电压极低会减缓核酸的迁移,从而导致小分子的扩散和分辨率过低(图13A)。另一方面,电压过高,会导致较差的分离效果和样品污点;有时,还会造成缓冲液过热,“微笑型条带”,甚至是样品变性(图13B)。
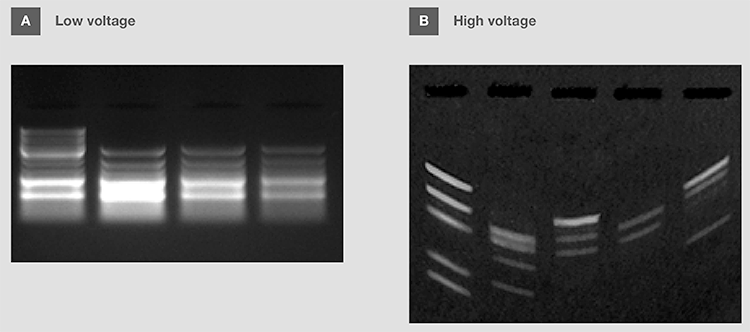
图13. 电压对DNA电泳的影响。 (A)低电压导致条带分辨率差和扩散。 (B)高电压导致“微笑型条带”。

某些情况下,可将温度探头连接至凝胶装置,以帮助控制缓冲液的冷却和加热。 电泳运行>2小时,缓冲液的冷却和再循环可改善样品分离。变性凝胶允许缓冲液加热至55°C,从而改善核酸单链形式,提升实验结果。
c.运行时间
凝胶长度、所用电压和样品中的分子大小决定电泳所需的时间。通常,电泳会持续到目的条带迁移至凝胶长度的40-60%。凝胶运行的特定时间,可观察到上样染料的相对位置,直至溴酚蓝染料已迁移约60%的凝胶长度和/或橙G染料已迁移80%的凝胶长度。此外,应监测运行时间,以确保样品或标准品中最小的分子不移出凝胶。注意,运行时间过短则不能完全分辨条带(图14)。含有示踪染料的DNA分子量标准可协助监测凝胶的运行,同时也可确保条带不被染料所遮盖。
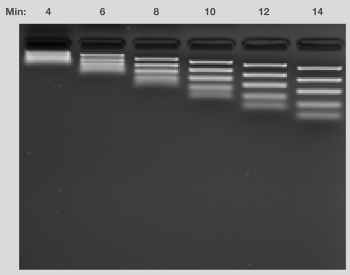
图14.运行时间对样品分离的影响。
4.在凝胶中可视化样品
凝胶运行完成后,需对样品进行可视化分析。由于普通照明环境下,核酸不可见,因此需要一种可视化的检测方法。如表9所述,可用方法在样品检测中,提供了不同的灵敏度和增益范围[6]。
a.荧光染色
现有的染色剂中,因荧光染料易于使用,灵敏度高,所以在样品检测中应用最为广泛(图15)。 当用适当波长激发时,染料发出可见光(即,荧光)。荧光强度与其和核酸结合的数量有关—这是检测和定量测定电泳中核酸的基础。
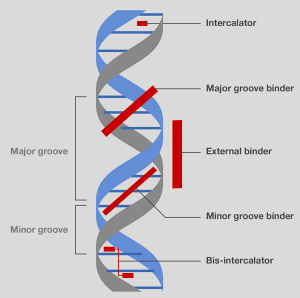
图15.不同类型的核酸结合染料。
溴化乙锭(EtBr) 染色时间短(约30分钟),灵敏度高(可检测到1 ng双链DNA/条带),为核酸电泳中的常用荧光染料。还可考虑另一种染料,原因如下:
诱变和处置: 溴化乙锭是高度诱变剂,所以荧光染料的 危险性较小,无需特殊处理,在核酸电泳中可提供安全工作流程(图16)。
灵敏度:灵敏度高于EtBr的荧光染料,适合检测少量样品(图16)。 因为有较少的碱基堆积,单链核酸的可视化通常需要更多样品和/或嵌入染料。 因此, 优先结合单链核酸的染料是RNA电泳样品检测的最佳选择。
紫外线损害:可用低能量蓝光(而非紫外光)来激发的荧光染料对核酸结构损伤较小,其灵敏度(图17A)不仅相同,甚至更高。 因此,电泳中使用蓝光激发可提高下游的成功应用,如克隆和测序(图17B)。

图16.特性增强的溴化乙锭替代品
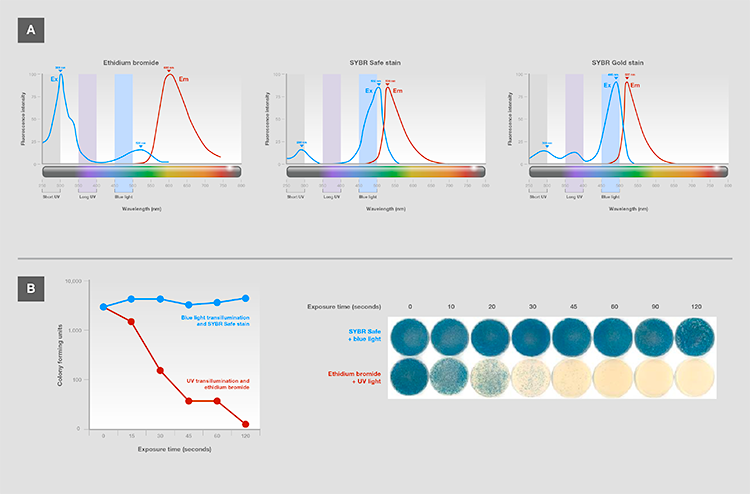
图17.(A)常见核酸染色的激发和发射光谱。最有效的波长被称为激发极大值。SYBR安全与SYBR金染料通过蓝光和较低的紫外光可最大程度激发。(B)用蓝光(针对SYBR安全染料)或紫外光(溴化乙锭)后的克隆效率,用于在电泳过程中显示克隆插入。用x-Gal培养皿上形成的蓝色菌落数量来衡量胶-纯化lacZ片段的克隆效率,其表明已将功能(未突变的)基因插入到载体中。
b.紫外阴影
在染料染色处,可通过称作紫外阴影的方法间接观察核酸,利用核酸[8]来吸收紫外线。该方法通常用于通过电泳分离和纯化寡核苷酸和RNA,这种情况下,简单检测已足够,和/或使用嵌入染料会影响下游的应用。为通过紫外阴影进行检测,需要纳克到微克的样品,并使用薄而透明的凝胶(如聚丙烯酰胺)以确保紫外线的吸收和透射。紫外阴影方法中,电泳后将凝胶从盒中取出(以便最大程度检测),用透明的塑料薄膜包裹后放置在UV-荧光薄层色谱板上。当凝胶至于紫外线辐射下时,核酸条带的吸收会在TLC板上投射阴影(图18)。将所需大小凝胶的阴影部分剪去,以作进一步处理。
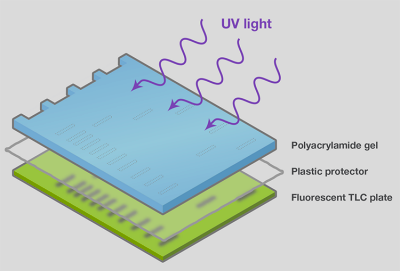
图18.紫外阴影使分离的片段显象。
5.记录凝胶
a.荧光成像技术
可视化后,核酸凝胶通常被存档记录下来,用于记录和分析电泳结果。如果样品被荧光染料染色,需特殊设备以适当的光源激发染料,使其显象并捕捉凝胶图像。激发光源在凝胶上,称为反射照明器(类似于手持式紫外线灯),或在凝胶下,称为透照器 (图19)。由于反射照明器上的光源位置较远,所以样品收到的能量较少。这样可以减少紫外线对核酸的损害,但也会降低凝胶条带的信号。另一方面,透照器可为条带提供更高的信号,但由于辐射接近凝胶,会增加紫外线造成的伤害。
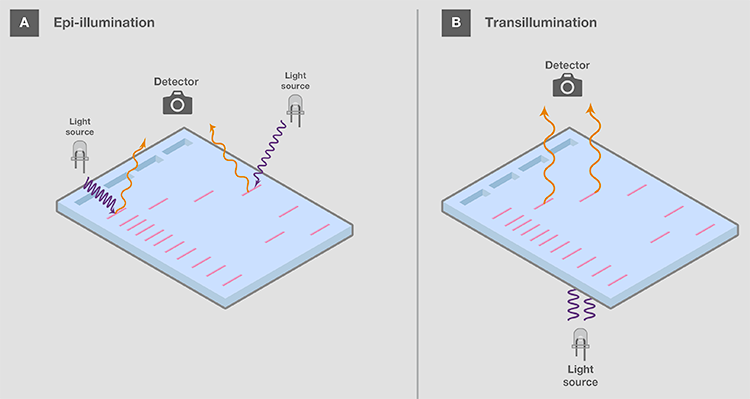
图19.反射照明器和透照器。
b.放射自显影法
在放射性核酸的电泳过程中,凝胶电泳后会暴露在x射线胶片上,这一过程即称为 放射自显影法。条带辐射的强度可用光密度测定来定量。
综上所述,核酸电泳工作流程采用多种步骤和试剂来分离和分析样品。为您的样品选择合适工具,并识别工作流程的优缺点,可显著提高分子生物学应用的电泳结果。
参考文献
1.Stellwagen NC (1998) DNA Gel Electrophoresis. In: Tietz D (editor), Nucleic Acid Electrophoresis (Springer Lab Manual). Heidelberg: Springer. pp 1–53.
2.Green MR, Sambrook J (2012) Analysis of DNA. In: Molecular Cloning: A Laboratory Manual (4th ed). Cold Spring Harbor: Cold Spring Harbor Laboratory Press. pp 81–156.